Frequently asked questions¶
Data filtering¶
Data filtering is an important step for deconvolution.
utilities/dataExplore.r -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-o PG0415-C
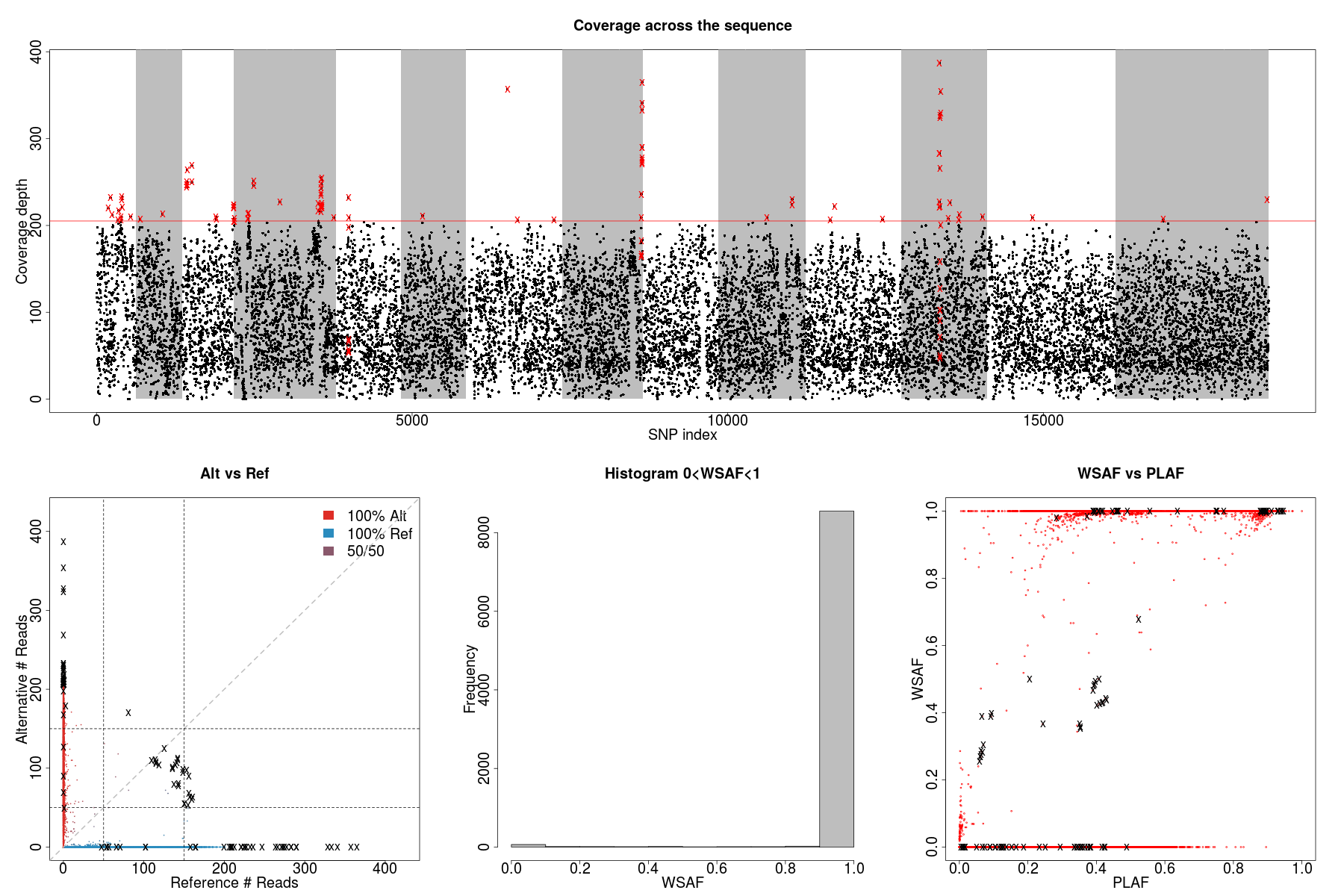 PG0415_data
PG0415_data
We observe a small number of heterozygous sites with high coverage (marked as crosses above), which can potentially mislead our model to over-fit the data with additional strains.
./dEploid -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-noPanel -o PG0415-CNopanel -seed 2
initialProp=$( cat PG0415-CNopanel.prop | tail -1 | sed -e "s/\t/ /g" )
./dEploid -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-panel data/exampleData/labStrains.eg.panel.txt \
-o PG0415-CNopanel \
-initialP ${initialProp} \
-painting PG0415-CNopanel.hap
utilities/interpretDEploid.r -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-dEprefix PG0415-CNopanel \
-o PG0415-CNopanel \
-ring
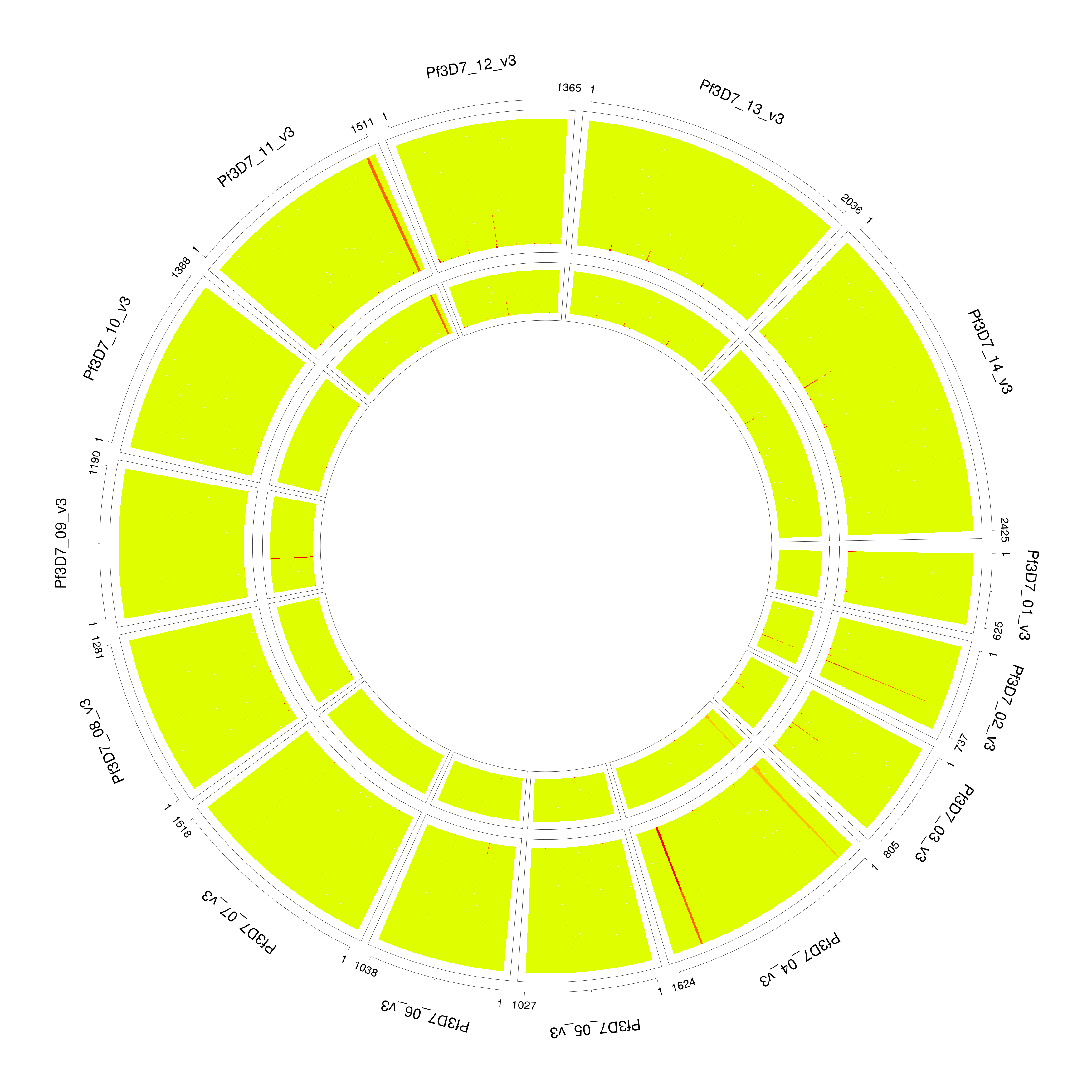 PG0415_noFilter
PG0415_noFilter
The data exploration utility utilities/dataExplore.r identifies a list of potential outliers. After filtering, we correctly identify the number of strains and proportion.
./dEploid -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-noPanel -o PG0415-CNopanel.filtered -seed 2 \
-exclude PG0415-CPotentialOutliers.txt
initialProp=$( cat PG0415-CNopanel.filtered.prop | tail -1 | sed -e "s/\t/ /g" )
./dEploid -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-panel data/exampleData/labStrains.eg.panel.txt \
-exclude PG0415-CPotentialOutliers.txt \
-o PG0415-CNopanel.filtered \
-initialP ${initialProp} \
-painting PG0415-CNopanel.filtered.hap
utilities/interpretDEploid.r -vcf data/exampleData/PG0415-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-dEprefix PG0415-CNopanel.filtered \
-o PG0415-CNopanel.filtered \
-exclude PG0415-CPotentialOutliers.txt \
-ring
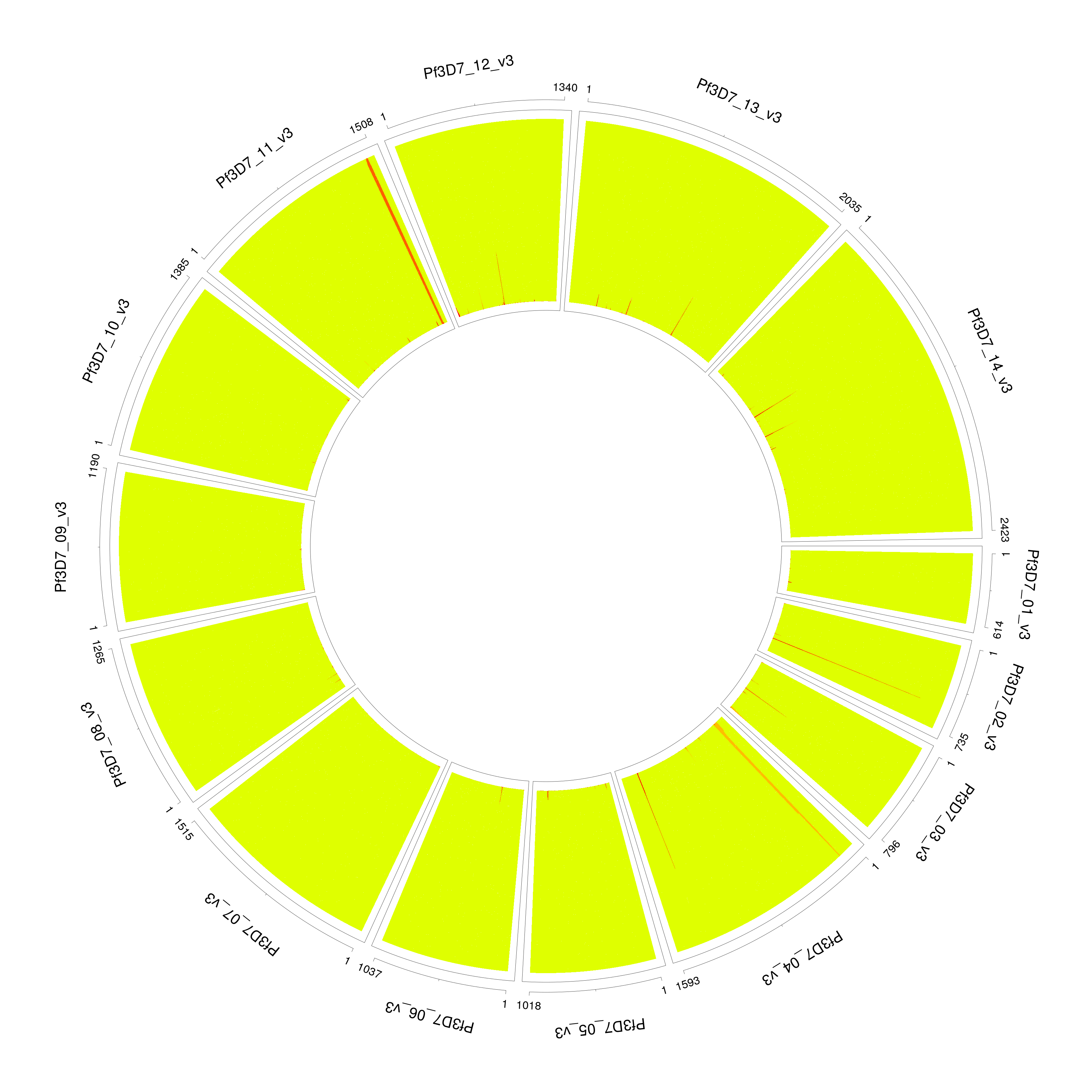 PG0415_filtered
PG0415_filtered
Over-fitting¶
For extremely unbalanced samples, DEploid tends to over-fit the minor strain with an additional component. We recommend adjusting the value of sigma for the prior to improve inference. In this example PG0400-C is a mixture of lab strains 7G8 and HB3 with mixing proportions of 95/5%. The parameter sigma takes value of 5 by default, which over fits the minor strain (see example 1), and with proportions 0.0276862, 0.945509 and 0.0267463. Example 1 paints the deconvolved strains (proportions in increasing order towards the centre) to the reference panel. We resolve the over-fitting issue by rerun this example, and set sigma with value of 10, it correctly infer the proportions as 0.0313755 and 0.968599 (see example 2). Note that the radius are not in scale with strain proportions.
./dEploid -vcf data/exampleData/PG0400-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-panel data/exampleData/labStrains.eg.panel.txt \
-o PG0400-Csigma5 -seed 2 -sigma 5 \
-exclude exclude.txt
initialProp=$( cat PG0400-Csigma5.prop | tail -1 | sed -e "s/\t/ /g" )
./dEploid -vcf data/exampleData/PG0400-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-panel data/exampleData/labStrains.eg.panel.txt \
-exclude exclude.txt \
-o PG0400-Csigma5 \
-initialP ${initialProp} \
-painting PG0400-Csigma5.hap
utilities/interpretDEploid.r -vcf data/exampleData/PG0400-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-dEprefix PG0400-Csigma5 \
-o PG0400-Csigma5 \
-exclude exclude.txt \
-reverseRing -transformP
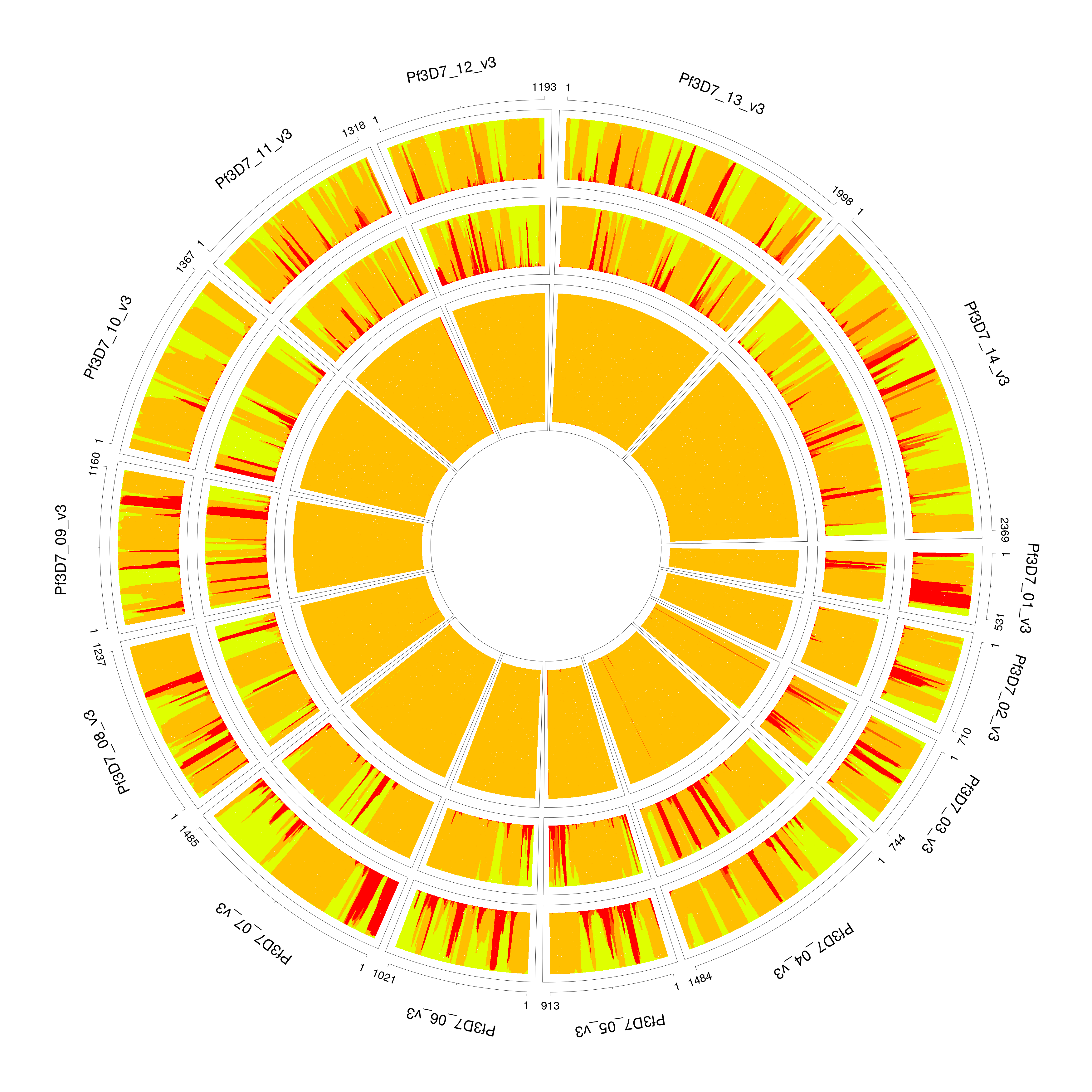 PG0400_sigma5
PG0400_sigma5./dEploid -vcf data/exampleData/PG0400-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-panel data/exampleData/labStrains.eg.panel.txt \
-o PG0400-Csigma10 -seed 2 -sigma 10\
-exclude exclude.txt
initialProp=$( cat PG0400-Csigma10.prop | tail -1 | sed -e "s/\t/ /g" )
./dEploid -vcf data/exampleData/PG0400-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-panel data/exampleData/labStrains.eg.panel.txt \
-exclude exclude.txt \
-o PG0400-Csigma10 \
-initialP ${initialProp} \
-painting PG0400-Csigma10.hap
utilities/interpretDEploid.r -vcf data/exampleData/PG0400-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-dEprefix PG0400-Csigma10 \
-o PG0400-Csigma10 \
-exclude exclude.txt \
-reverseRing -transformP
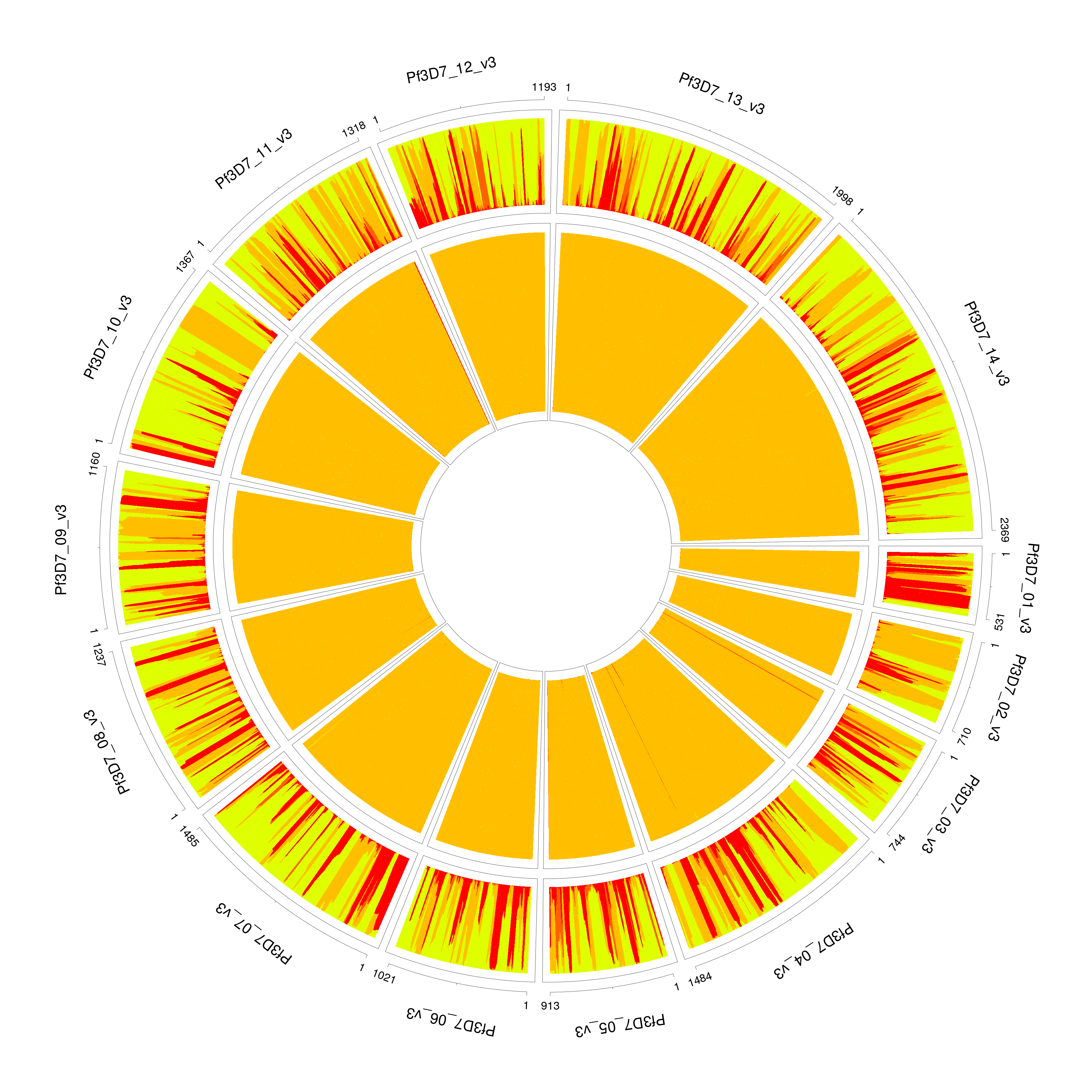 #PG0400_sigma10
#PG0400_sigma10Benchmark¶
Please refer to our paper Zhu et.al (2017) section 3 Validation and performance for benchmarking inference results on number of strains, proportions and haplotype quality.
For the enhanced version – DEploid-IBD, we compared our results against Zhu et.al (2017), and conducted more experiments and validations Zhu et.al (2019).