How it works?¶
Program parameters and options¶
Mostly used¶
- -vcf [file]
- File path of the isolate vcf. Assume all variants are
PASSin theQUALcolumn, the VCF file also reqires theADfield.
Note
In the current implementation, DEploid only take the first sample in the VCF file. DEploid DO NOT handle multi-allelic variants, nor indels. The FILTER column will not be used.
- -plaf [file]
- File path of population level allele frequencies (tab-delimited plain text file), for example
| CHROM | POS | PLAF |
|---|---|---|
| Pf3D7_01_v3 | 93157 | 0.0190612159917058 |
| Pf3D7_01_v3 | 94422 | 0.135502358766423 |
| Pf3D7_01_v3 | 94459 | 0.156294363760064 |
| Pf3D7_01_v3 | 94487 | 0.143439298925837 |
- -panel [file]
- File path of the reference panel (tab-delimited plain text file), for example
| CHROM | POS | 3D7 | Dd2 | Hb3 | 7G8 |
|---|---|---|---|---|---|
| Pf3D7_01_v3 | 93157 | 0 | 0 | 0 | 1 |
| Pf3D7_01_v3 | 94422 | 0 | 0 | 0 | 1 |
| Pf3D7_01_v3 | 94459 | 0 | 0 | 0 | 1 |
| Pf3D7_01_v3 | 94487 | 0 | 0 | 0 | 1 |
- -noPanel
- Use population level allele frequency as prior.
Warning
Flags -panel and -noPanel should not be used together.
- -exclude [file]
- File path of sites to be excluded (tab-delimited plain text file).
- -o [string]
- Specify the file name prefix of the output.
- -k [int]
- Number of strain (default value 5).
- -seed [int]
- Random seed.
- -nSample [int]
- Number of MCMC samples (default value 800).
- -rate [int]
- MCMC sample rate (default value 5).
- -burn [float]
- MCMC burn rate (default value 0.5).
- -ibd
- Use IBD segment to infer the proportion, then infer the haplotype (see Pf3k work-flow for more details).
- -painting [file]
- Paint the posterior probability of the given haplotypes.
- -inbreeding
- Calculate the inbreeding probabilities.
- -initialP [float …]
- Initialize proportions.
- -ibdPainting
- IBD painting, compute posterior probabilities of IBD configurations of given strain proportions. This option must be used with flags -initialP.
- -h , -help
- Help.
- -v , -version
- DEploid version.
- -vcfOut
- Save final halpotypes into a VCF file.
You may also try¶
- -ref [file]
- File path of reference allele count (tab-delimited plain text file).
Note
In early dEploid versions (prior to v0.2-release), allele counts extracted from the vcf file are placed in two files, and parsed by flags -ref [file] and -alt [file]. Tab-delimited plain text for input. First and second columns record chromosome and position labels respectively. Third columns records the reference allele count or alternative allele count. For example,
| CHROM | POS | PG0390.C |
|---|---|---|
| Pf3D7_01_v3 | 93157 | 85 |
| Pf3D7_01_v3 | 94422 | 77 |
| Pf3D7_01_v3 | 94459 | 90 |
| Pf3D7_01_v3 | 94487 | 79 |
- -alt [file]
- File path of alternative allele count (tab-delimited plain text file).
| CHROM | POS | PG0390.C |
|---|---|---|
| Pf3D7_01_v3 | 93157 | 0 |
| Pf3D7_01_v3 | 94422 | 0 |
| Pf3D7_01_v3 | 94459 | 0 |
| Pf3D7_01_v3 | 94487 | 0 |
Warning
Flags -ref and -alt should not be used with -vcf.
- -forbidUpdateProp
- Forbid MCMC moves to update proportions.
- -forbidUpdateSingle
- Forbid MCMC moves to update single haplotype.
- -forbidUpdatePair
- Forbid MCMC moves to update pair haplotypes.
- -exportPostProb
- Save the posterior probabilities of the final iteration of all strains.
- -miss [float]
- Miss copying probability.
- -recomb [float]
- Constant recombination probability.
- -p [int]
- Output precision (default value 8).
- -c [float]
- Specify scaling parameter c, which reflects how much data is available (default value 100.0).
- -G [float]
- Specify scaling parameter for genetic map (default value of 20.0).
- -sigma [float]
- Specify the variance parameter for proportion estimation (default value of 5.0).
- -ibdSigma [flat]
- Specify the variance parameter for proportion estimation when IBD method is used (default value of 20.0).
- -initialHap [file]
- Specify initial haplotypes of deconvolution.
R utilities¶
Flags -vcf, -plaf, -ref, -alt, -exclude, -o usage are the same as DEploid. Additionally, we have the following flags:
- -dEprefix [string]
- Prefix of
DEploidoutput. - -inbreeding
- Painting haplotype inbreeding posterior probabilities.
- -ADFieldIndex
- The index of
ADfield (2 by default). - -filter.threshold [float]
- Filtering threshold (0.995 by default).
- -filter.window [int]
- Filtering window (10 by default).
- Produce figures in pdf rather than png.
- -ibd
- Produce figures for IBD process.
- -ring
- Produce circular genome plots for WSAF and haplotype posterior painting probabilities.
Example of data exploration¶
Use our data exploration tools to investigate the data.
$ utilities/dataExplore.r -vcf data/exampleData/PG0390-C.eg.vcf.gz \
-plaf data/exampleData/labStrains.eg.PLAF.txt \
-o PG0390-C
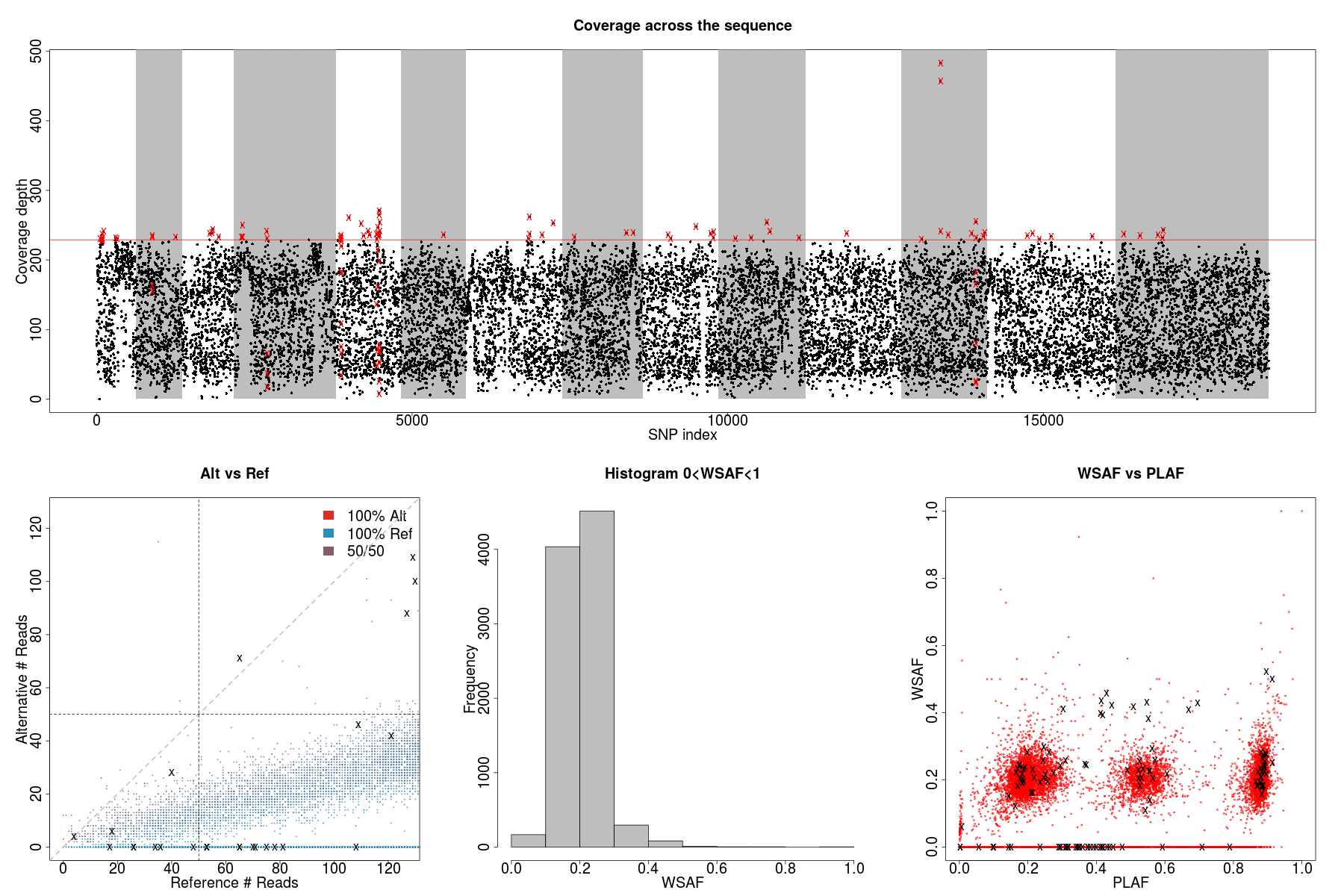
- Figure on the top plot total allele counts across all markers. We use the threshold (red line) to identify markers with extremely high allele counts. Red crosses indicate markers that are filtered out.
- Figure on the left plots the alternative allele count against the reference allele count. As P. falciparum genomes are haploid, in clonal samples, one woule expect to see either alternative or reference allele at any sites. Heterozygous sites are indications of mixed infection.
- Figure in the middle is the histogram of the allele frequency within sample. Note that we exclude markers with WSAF strictly equal to 0s and 1s in the histogram.
- Figure on the right show allele frequency within sample, compare against the population average.